Differential expression with DESeq2
Learning Objectives
- Install the DESeq2 package for use in R and RStudio
- Create a sample sheet for your differential expression analysis
- Import transcript abundance data from Salmon into a DESeq2 object
- Run differential expression analysis with DESeq2
- Assess replicate and sample groups (PCA plots and hierarchical clustering)
- Extract and visualise results (MA plots, scatter plots, volcano plots)
- Search for enriched functional annotations in differentially expressed genes (GO terms, gProfiler2)
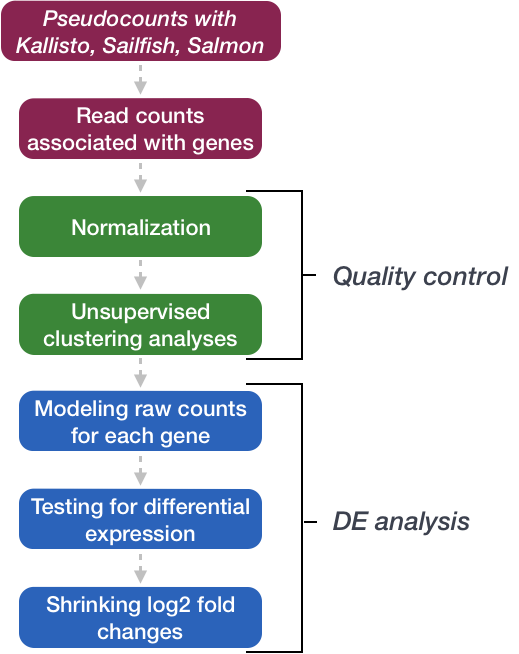
1. Getting Started
Differential expression (DE) analysis is commonly performed downstream of RNA-seq data analysis and quantification. We use statistical methods to test for differences in expression of individual genes between two or more sample groups. In this lesson, we will use the statistical programming language R and the DESeq2 package, specifically designed for differential expression analysis.
R and RStudio
R is an extremely powerful programming language for working with large datasets, applying statistical tests and creating publication ready graphics. RStudio is an Integrated Development Environment (IDE) for R which provides a graphical and interactive environment for R programming.
We recommend that you understand the basics of R and RStudio and follow at least the getting started and R and RStudio lessons in our introduction to R workshop. This will also guide you through how to install R and RStudio on your own machine.
You can choose to run RStudio on your own laptop or log into our RStudio server on the bioinformatics servers.
Installing DESeq2
DESeq2 is part of the Bioconductor repository of biology specific R packages. You can install DESeq2 using the Bioconductor manager with the code below in the R Console. We will also use several other R libraries.
## Install Bioconductor packages
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("DESeq2")
BiocManager::install("tximeta")
BiocManager::install("apeglm")
BiocManager::install("clusterProfiler")
BiocManager::install("org.Hs.eg.db")
## Install CRAN packages
install.packages("tidyverse")
install.packages("RColorBrewer")
install.packages("gprofiler2")
install.packages("pheatmap")Create a project
Use the project drop-down menu at the top right to create a new project called Differential expression workshop or something similar and choose an appropriate working directory.
Input data
DESeq2 works with matrices of read counts per gene for multiple samples. In the past we used read counting software like HTSeq-count or featureCounts to quantify counts of aligned reads (e.g. from STAR) over exons for each gene model. The standard practice now is to use pseudocounts from tools like Salmon which do a much better job at estimating expression levels by:
- Correcting for sequencing biases e.g. GC content
- Correcting differences in individual transcript lengths
- Including reads that map to multiple genes
- Producing much smaller output files than aligners
DESeq2 requires non-normalised or “raw” count estimates at the gene-level for performing DE analysis. We will use the R package tximport to import read counts and summarise transcript abundance estimates for each gene.
In the previous lessons we generated tables of transcript abundances
(quant.sf) for each sample with Salmon, using a reduced set
of RNA-seq sequencing reads. As input to DESeq2 we will use similar
tables generated from the full dataset.
Download and extract the data folder:
download.file("https://bifx-core3.bio.ed.ac.uk/training/RNA-seq_analysis/salmon.tar.gz","salmon.tar.gz",method="curl",extra=c("--insecure"))
untar("salmon.tar.gz")Experimental design
We will also include a further two samples from the original publication which correspond to a MOV10 knock-down cell line using siRNA. We will use this data to investigate changes in transcription upon perturbation of MOV10 expression relative to the control (irrelevant siRNA) experiment.
| Dataset | Description |
|---|---|
| Control_1 | Control, replicate 1 |
| Control_2 | Control, replicate 2 |
| Control_3 | Control, replicate 3 |
| MOV10_OE_1 | MOV10 over-expression, replicate 1 |
| MOV10_OE_2 | MOV10 over-expression, replicate 2 |
| MOV10_OE_3 | MOV10 over-expression, replicate 3 |
| MOV10_KD_2 | MOV10 knock-down, replicate 2 |
| MOV10_KD_3 | MOV10 knock-down, replicate 3 |
MOV10 is an RNA helicase reported to associate with FMR1, a protein found in the brain and linked to fragile X syndrome.
Notes on experimental design
DESeq2 and other differential expression tools perform statistical analysis by comparing mean expression levels between sample groups. Accurate mean estimates can only be achieved with precise estimates of the biological variation between independent samples.
In DE experiments, increasing the number of biological replicates is generally more desirable than increasing read depth in single samples, unless you are particularly interested in rare RNAs. DESeq2 will not work if a sample group only has one replicate and it is recommended to have at least 3.
If you have technical replicates in your experimental design, these should be merged into one sample to represent a single biological replicate.
We also need to ensure (as much as possible) that any effect we see between groups can be attributed to differences in biology rather than confounding factors in the design or batch effects introduced at the library preparation stage.
Further Learning
Read this document on experimental design considerations in differential expression analysis.
The DESeq2 method
DESeq2 performs statistical analysis of un-normalised raw/estimated read count data per gene. It uses a median of ratios normalisation method to account for differences in sequencing depth and RNA composition between samples.
Count data is modeled using a generalised linear model based on the negative binomial distribution, with a fitted mean and a gene-specific dispersion parameter which describes the relationship between variance in the count data and the observed mean.
The model coefficient represents the change in mean between sample groups giving us log2 fold change values per gene. By default, DESeq2 performs the Wald test to test for significant changes in gene expression between sample groups and generate p-values which are then adjusted for multiple testing.
DESeq2 has internal methods for:
- Estimating size factors (sample normalisation)
- Estimating dispersions
- Fitting the negative binomial GLM (log2 fold changes)
- Filtering outliers and low count genes
- Statistical tests between sample groups (p-values)
- Multiple testing correction (FDR adjusted p-value)
Comprehensive tutorials
This is a lightweight introduction to differential expression analysis. For a comprehensive overview of the DESeq2 method, functionality and complex experimental designs, check out the following resources:
Key points:
- Setup R and RStudio and install required packages
- Download the required datasets
- Understand considerations for experimental design
- Understand an overview of the DESeq2 methodology
2. Create a sample file
The first step in our analysis is to create a tab separated file of
sample IDs and metadata. This already exists in the folder you
downloaded earlier but you would normally create this manually. We can
then import the sample sheet to R with the read_tsv()
function from readr.
library(tidyverse)
ss<-read_tsv("salmon/samples.tsv",col_names = T,col_types = "fff")
ssKey points:
- The sample file should contain metadata on each sample
- It is important to include all known variables that could confound results or explain technical variance
3. Importing count data with tximeta
We will start by importing the count data for each sample into R. We will use the package tximeta to read the Salmon output files and create a matrix of read counts for each gene. This package automatically recognises if your count data comes from a commonly used genome and will download and add annotation files. Alternatively, you can use tximport.
library(DESeq2)
library(tximeta)
## tximeta expects a table with at least 2 columns (names and files). We will also add the Group and Replicate.
coldata<-ss %>% dplyr::select(names=Sample,condition=Group,replicate=Replicate) %>%
mutate(files=file.path("salmon", ss$Sample, "quant.sf"))## Read in Salmon counts with tximeta - this creates an object of type summarizedExperiment
se<-tximeta(coldata)## importing quantifications## reading in files with read_tsv## 1 2 3 4 5 6 7 8
## found matching transcriptome:
## [ Ensembl - Homo sapiens - release 106 ]
## loading existing EnsDb created: 2022-06-15 11:06:58
## loading existing transcript ranges created: 2022-06-15 11:07:06se## class: RangedSummarizedExperiment
## dim: 252199 8
## metadata(6): tximetaInfo quantInfo ... txomeInfo txdbInfo
## assays(3): counts abundance length
## rownames(252199): ENST00000631435 ENST00000415118 ... ENST00000619896
## ENST00000582918
## rowData names(11): tx_id tx_biotype ... tx_is_canonical tx_name
## colnames(8): Control_1 Control_2 ... MOV10_OE_2 MOV10_OE_3
## colData names(3): names condition replicateThe summarizedExperiment object in R contains a matrix of count data with sample names as columns and features as rows. It can also hold metadata and annotation data. At the moment our counts are per-transcript but DESeq2 works on a per-gene level. We can summarise our counts into genes with the summarizeToGene function in tximeta and tximport.
gse<-summarizeToGene(se)## loading existing EnsDb created: 2022-06-15 11:06:58## obtaining transcript-to-gene mapping from database## loading existing gene ranges created: 2022-06-15 11:09:01## summarizing abundance## summarizing counts## summarizing lengthgse## class: RangedSummarizedExperiment
## dim: 63345 8
## metadata(6): tximetaInfo quantInfo ... txomeInfo txdbInfo
## assays(3): counts abundance length
## rownames(63345): ENSG00000000003 ENSG00000000005 ... ENSG00000289718
## ENSG00000289719
## rowData names(10): gene_id gene_name ... entrezid tx_ids
## colnames(8): Control_1 Control_2 ... MOV10_OE_2 MOV10_OE_3
## colData names(3): names condition replicateCompare the number of rows in se and gse.
You can access data within a summarizedExperiment object with
accessor functions such as colData(),
rowRanges() and assay().
Now that we have per-gene count data in a
summarizedExperiment object, we can import this into DESeq2. We
need to supply the gse object we have just created as well
as a design.
The simplest design is just to compare our samples by the condition
column (Control, MOV10_KD, MOV10_OE). The design is a formula
in R so is preceded with the ~ character.
Further Learning
Have a look at these resources for advice on complex experimental designs (e.g. multiple variables of interest, interaction terms, time-course analysis:
- DESeq2 manual
- Time-course analysis example
- Likelihood ratio test for measuring changes across multiple sample groups at once.
Key points:
- Import pseudo-count data into R with tximeta or tximport
- DESeq2 expects gene-level counts
- Summarise transcript counts to gene counts
4. Creating a DESeq object and running DESeq
## Create the DESeq dataset object
dds <- DESeqDataSet(gse, design = ~ condition)## using counts and average transcript lengths from tximetadds## class: DESeqDataSet
## dim: 63345 8
## metadata(7): tximetaInfo quantInfo ... txdbInfo version
## assays(3): counts abundance avgTxLength
## rownames(63345): ENSG00000000003 ENSG00000000005 ... ENSG00000289718
## ENSG00000289719
## rowData names(10): gene_id gene_name ... entrezid tx_ids
## colnames(8): Control_1 Control_2 ... MOV10_OE_2 MOV10_OE_3
## colData names(3): names condition replicateThe condition column is represented in R as a factor, or categorical variable, which has levels.
levels(dds$condition)## [1] "Control" "MOV10_KD" "MOV10_OE"By default, the levels are set in alphabetical order and DESeq2 will always assume that the first level is your control group to which it will compare datasets. In this case we are okay, otherwise you will need to relevel your condition column or explicitly reference the condition comparisons of interest in your results (see below).
Let’s create a list of comparisons, known as contrasts, that we want to look at. Here we will put our base-level or control sample last. We will also set a few other variables to help name our outputs.
contrasts<-list(c("condition","MOV10_OE","Control"),
c("condition","MOV10_KD","Control"))
## As well as contrasts, DESeq also uses coefficients to name results. We can create these from our specified contrasts
coefficients<-contrasts %>% map(~paste(.x[1],.x[2],"vs",.x[3],sep = "_")) %>% unlist()
## Simple project ID
project="MOV10"
## Labels for QC plots - we can add all possible confounding factors from our colData
labels= c("condition","replicate")
## Merge colData into label names in a data frame
cnames <- colData(dds) %>%
as_tibble() %>%
unite(all_of(labels), col = label, sep = "-") %>%
pull(label)
## Thresholds for p-value and fold change to filter and summarise results later
padj_thresh = 0.05
l2fc_thresh = 0 ## Just include all significant genes hereNow we are ready to run DESeq. The DESeq function has internal methods to:
- Estimate size factors to normalise gene counts per sample
- Estimate gene-wise dispersions to measure variance in the dataset
- Shrink gene-wise dispersions to improve the dispersion estimates
- Fit a negative binomial statistical model to the data
- Perform statistical testing with the Wald Test or Likelihood Ratio Test
## run DESeq
dds <- DESeq(dds)## estimating size factors## using 'avgTxLength' from assays(dds), correcting for library size## estimating dispersions## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## fitting model and testing## save dds object as a file - saveRDS can save R objects
saveRDS(dds,file =paste0(project,".dds.RDS"))Plot dispersions
It can be useful to plot the gene-level dispersion estimates to ensure the DESeq model is right for your data. You should find that dispersion is generally lower for genes with higher read counts, that final dispersion levels have been shrunk towards the fitted model and that a few outliers exist which have not been shrunk. If the red line is not a good generalisation for your data then DE analysis with DESeq2 may not be appropriate.
#Plot dispersions
plotDispEsts(dds, main="Dispersion plot")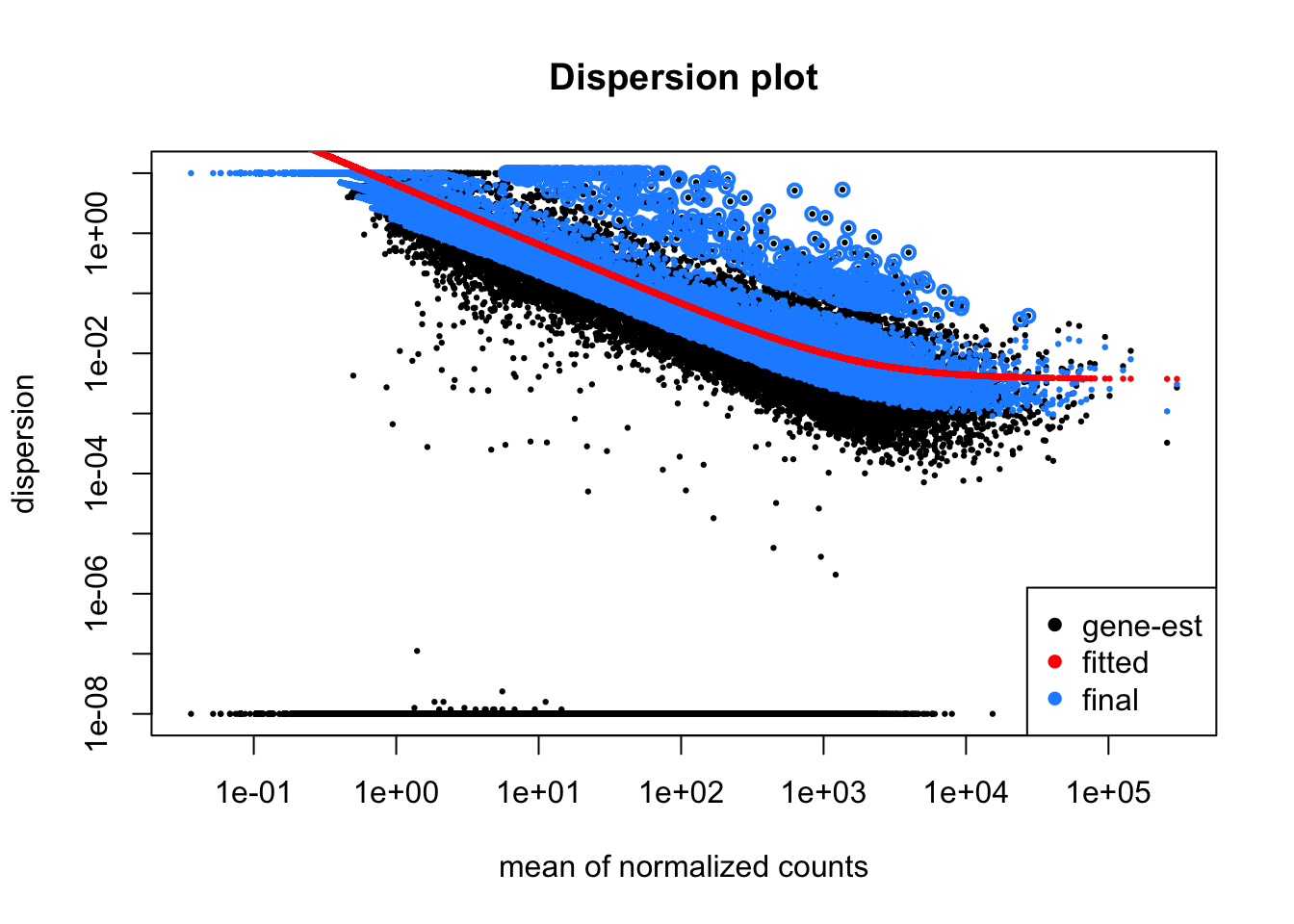
Key points:
- Create a DESeq dataset from count data
- Understand the DESeq design parameter
- Run the
DESeqfunction and understand the internal steps - Plot dispersions to assess the fitted model
5. DESeq quality control
Before we look at the results of differential expression tests we
first want to perform some quality control by visualising and assessing
the entire dataset. The raw counts are not optimal for visualisation and
clustering so we will apply a regularised log transformation
which reduces the bias from genes with extremely low and high counts.
The rld() function can take a while to run with large
datasets.
## Log transformed data
rld <- rlog(dds, blind=F)
saveRDS(rld,file =paste0(project,".rld.RDS"))Further Learning
You can read more on DESeq2 data transformations here.
Heatmap of Sample Distances
We can use our transformed data to perform sample clustering. Here,
we calculate sample distances with the dist()
function applied to the transpose of the transformed count matrix. A
heatmap of this distance matrix gives us an overview of similarities and
dissimilarities between samples.
We have to provide a hierarchical clustering hc to the
heatmap function based on the sample distances, or else the heatmap
function would calculate a clustering based on the distances between the
rows/columns of the distance matrix.
library("pheatmap") # heatmap plotting package
library("RColorBrewer") # colour scales
sampleDists <- dist(t(assay(rld)))
sampleDistMatrix <- as.matrix(sampleDists)
rownames(sampleDistMatrix) <- as.list(cnames)
colnames(sampleDistMatrix) <- as.list(cnames)
cols <- colorRampPalette( rev(brewer.pal(9, "Blues")) )(255) ## Set a colour pallette in shades of blue
pheatmap(sampleDistMatrix,
clustering_distance_rows=sampleDists,
clustering_distance_cols=sampleDists,
col=cols)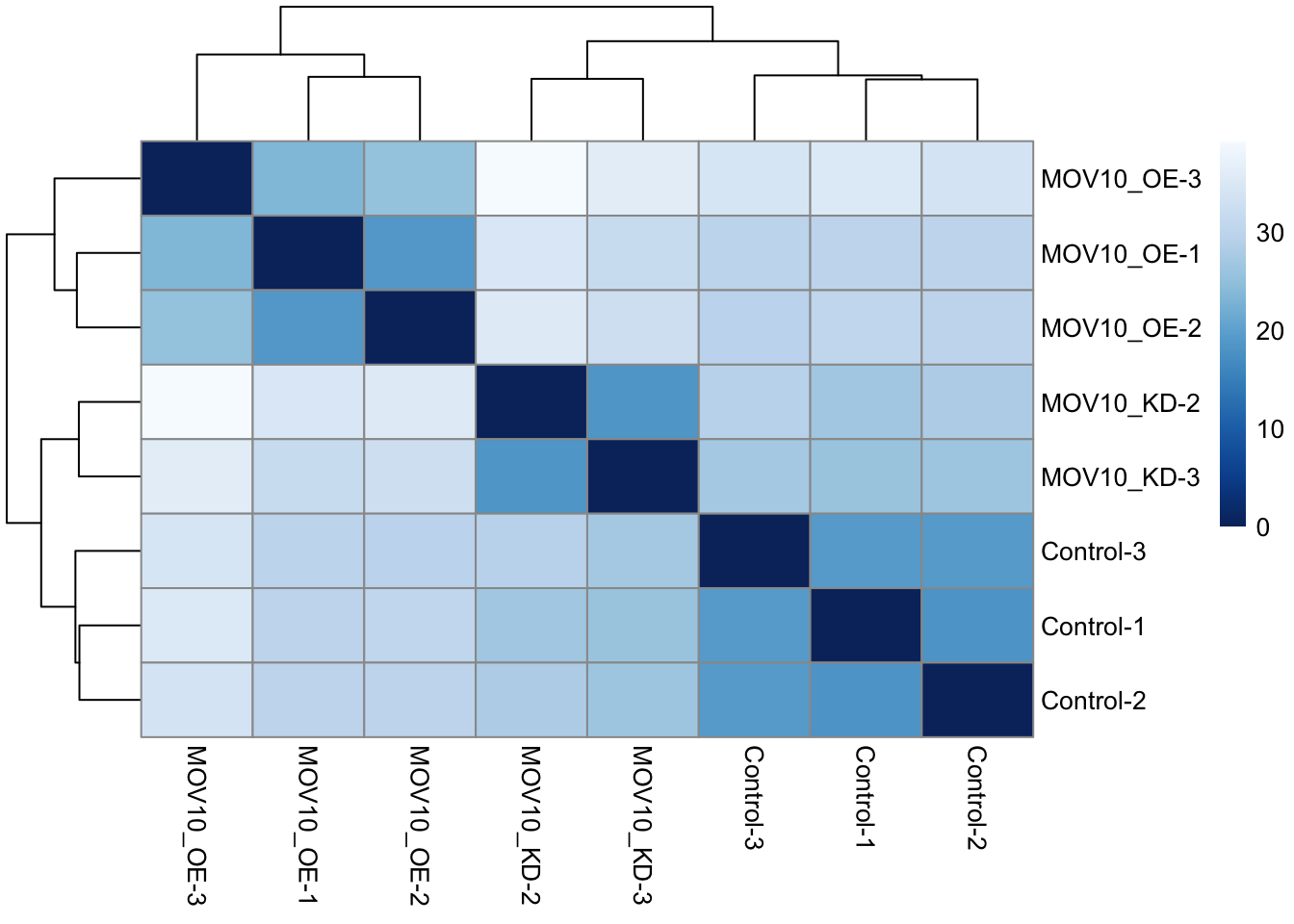
Principle Component Analysis
Another way to visualize sample-to-sample distances is a principal components analysis (PCA). In this method, the data points (here, the samples) are projected onto a 2D plane such that they spread out in the two directions that explain most of the variation in the data.
The x-axis is the direction that separates the data points the most. The values of the samples in this direction are written PC1 (principle component 1). The y-axis is a direction that separates the data the second most, PC2. The percent of the total variance that is contained in each direction is printed on the axis label. Note that these percentages do not add to 100%, because there are more dimensions that contain the remaining variance.
We expect to see our samples divide by their biological condition or some other source of variation that we are aware of (e.g. sex, cell type, batches of library preparation etc). If you do not see your samples separating by your variable of interest you may want to plot out PC3 and PC4 to see if it appears there. If there is a large amount of variance introduced by other factors or batch effects then you will need to control for these in your experimental design. See the DESeq2 vignette for more details.
We will run the PCA analysis with the DESeq2 command
plotPCA().
## Principle component analysis - get the PCA data
plotPCA(rld, intgroup=c(labels[1],labels[length(labels)]))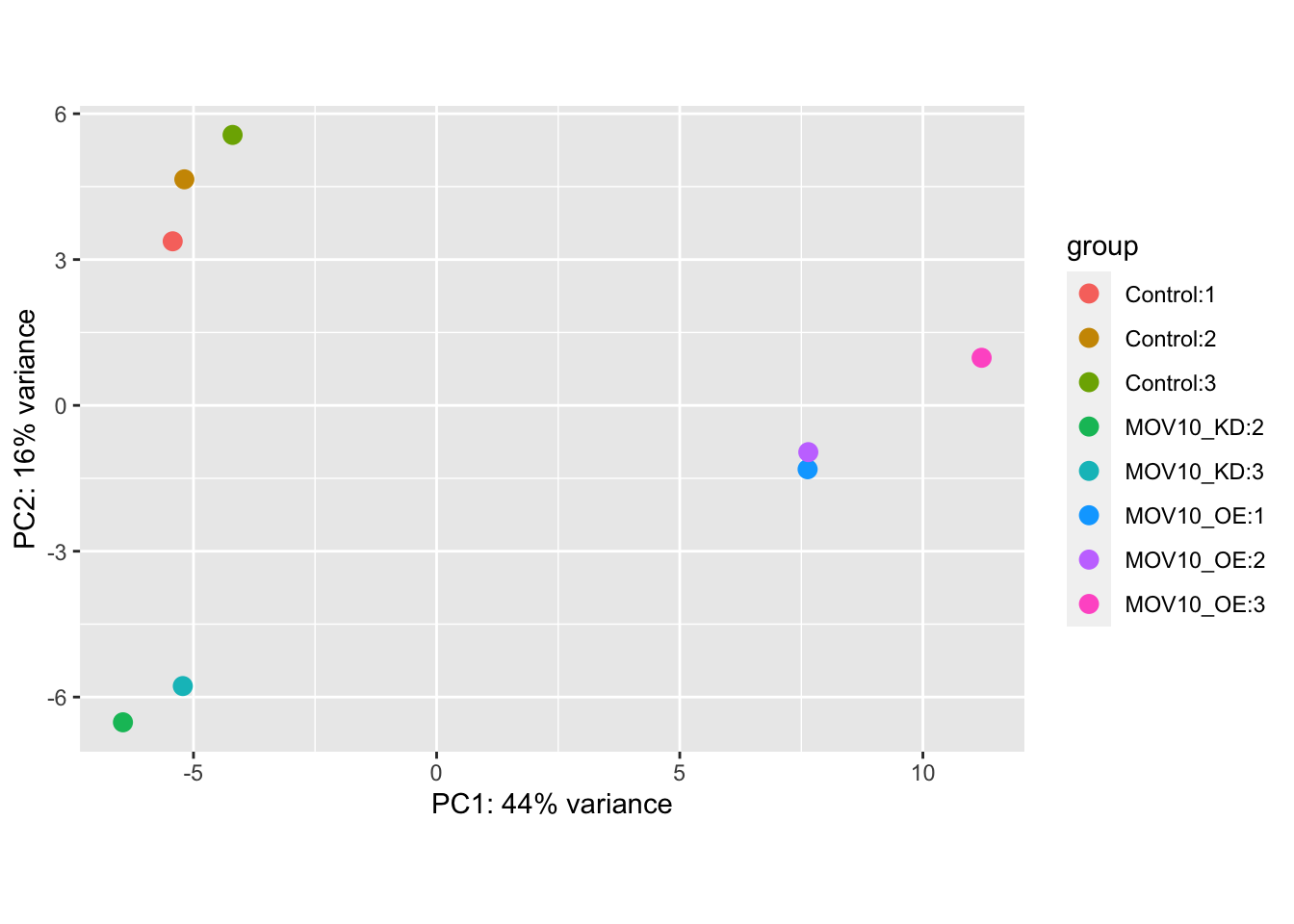
If we don’t like the default plotting style we can ask
plotPCA to return the data only and create our own custom
plot. Below, we use ggplot, a sophisticated plotting
package in R. For more on ggplot, refer to our introduction to R
workshop.
## Principle component analysis - get the PCA data
pca<-plotPCA(rld, intgroup=c(labels[1],labels[length(labels)]),returnData=T)
## Plot with ggplot
ggplot(pca,aes(PC1,PC2,colour=condition,shape=replicate)) +
geom_point(size=3) +
theme_bw() +
theme(legend.key = element_blank()) +
xlab(paste("PC1:",round(attr(pca,"percentVar")[1]*100),"%")) +
ylab(paste("PC1:",round(attr(pca,"percentVar")[2]*100),"%"))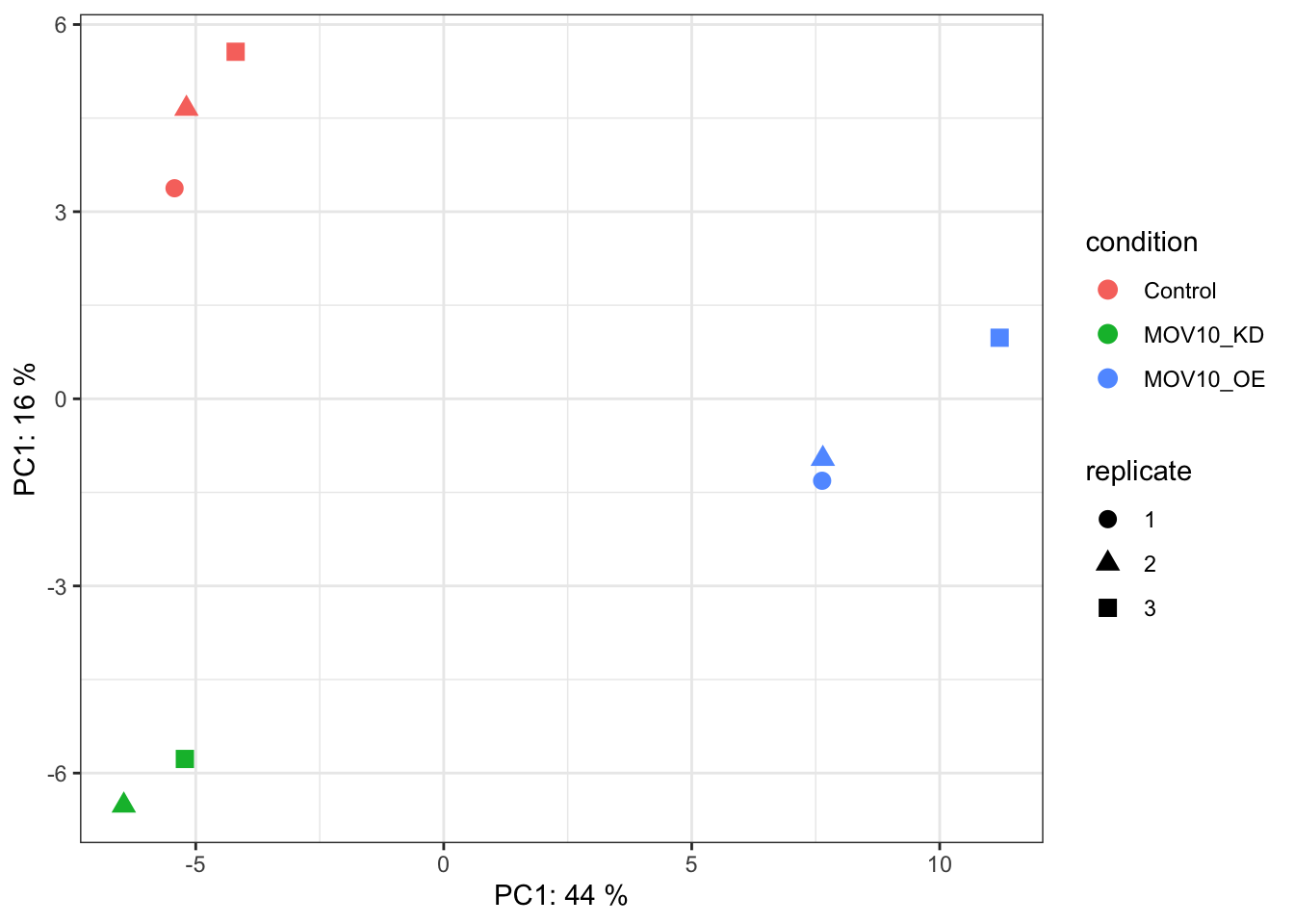
Heatmap of genes with the largest variance
It may also be useful to take an initial look at the genes with the highest amount of variation across the dataset. We should expect to see some genes which appear to be differentially expressed between sample groups.
## Get the top 20 genes after ordering the rld counts by variance
topVarGenes <- head(order(-rowVars(assay(rld))),20)
## Create a matrix from these genes only
mat <- assay(rld)[topVarGenes, ]
anno<-as.data.frame(colData(dds)[,labels])
pheatmap(mat,cluster_rows = F,cluster_cols = F,show_rownames = T,scale="row",annotation_col = anno)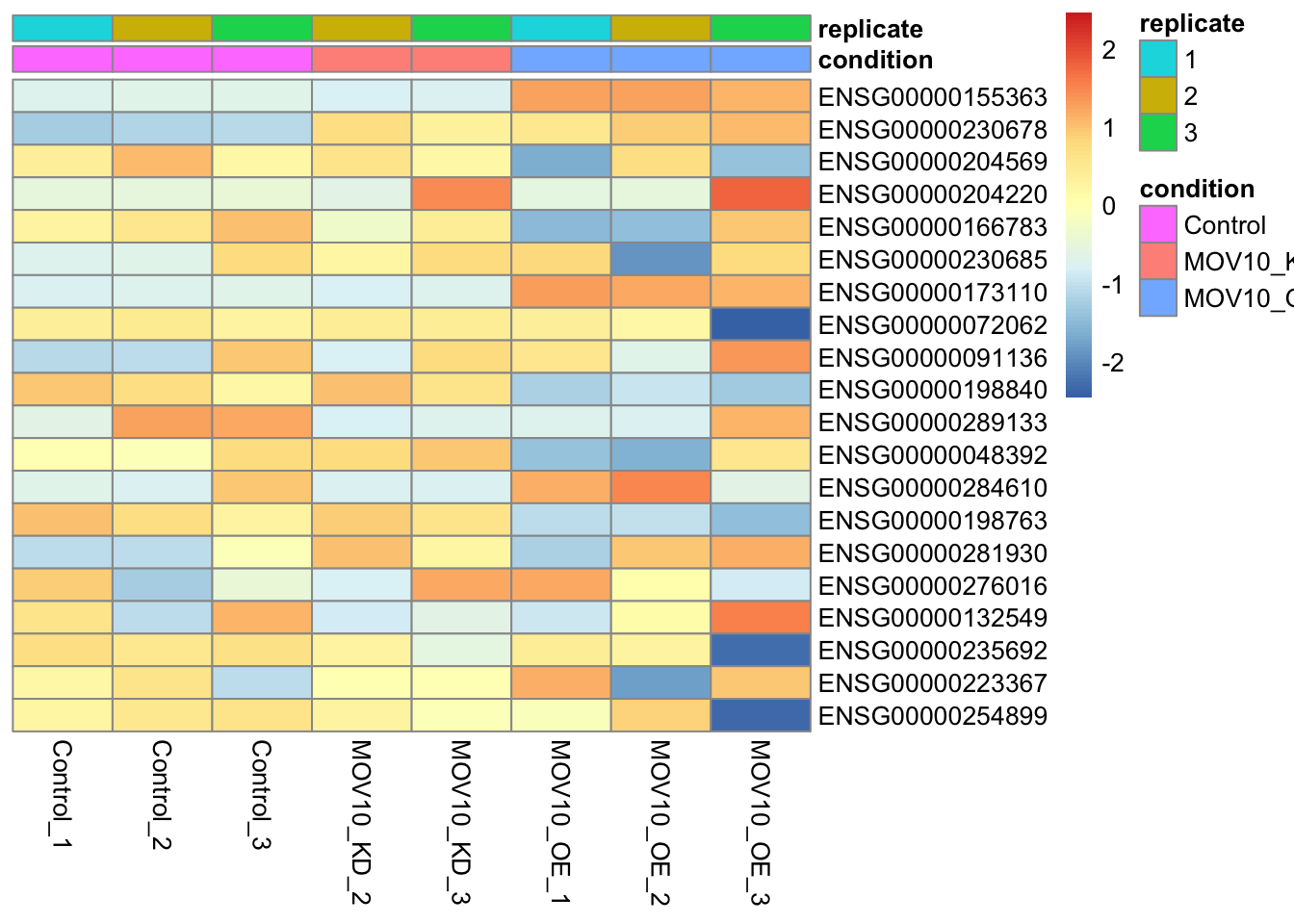
Discussion
Can you guess which gene has the Ensembl identifier ENSG00000155363?
Plot Individual Gene Counts
In certain cases like ours, where we know the expression levels of particular genes should change between sample groups, we may want to plot individual gene counts. Let’s plot normalised gene counts for the MOV10 gene (ENSG00000155363).
## Get normalised counts for a single gene
gene="ENSG00000155363"
geneData <- plotCounts(dds, gene=gene, intgroup=labels, returnData=TRUE,normalized = T)
## Plot with ggplot
ggplot(geneData, aes_string(x=labels[1], y="count",fill=labels[length(labels)])) +
scale_y_log10() +
geom_dotplot(binaxis="y", stackdir="center") +
theme_bw() +
theme(legend.key = element_blank()) +
ggtitle(gene) +
theme(axis.text.x = element_text(angle = 90, hjust = 1))## Bin width defaults to 1/30 of the range of the data. Pick better value with `binwidth`.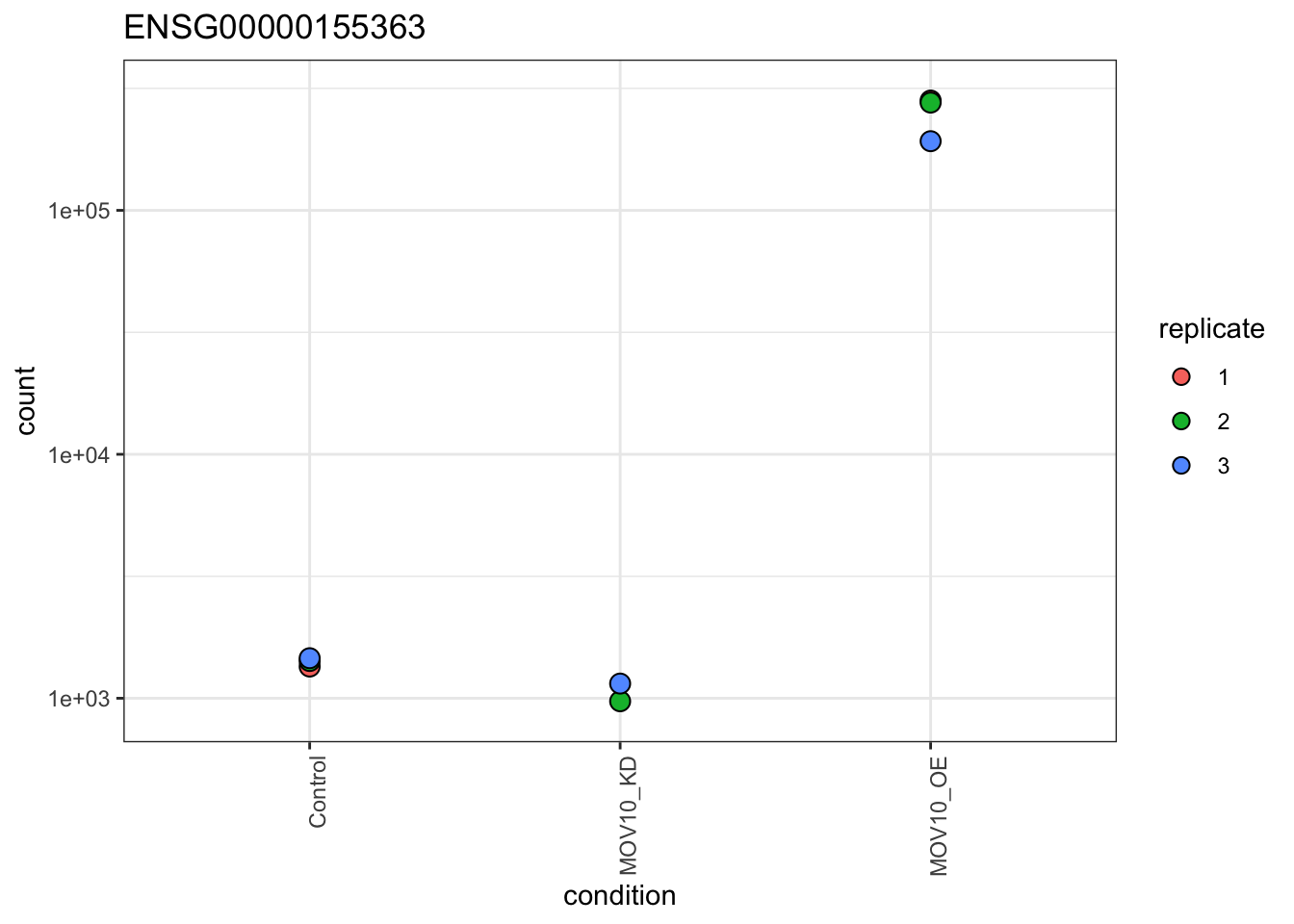
Key points:
- Transform count data for summary plots
- Create summary plots and assess for QC
- PCA
- Clustering by sample distance
- Heatmaps of gene counts
- Individual gene counts
6. Extract DESeq2 results
If we are happy with our QC assessment we can retrieve results from the DESeq object and visualise fold changes between specific comparisons.
DESeq2 has a results() function which by default will
print the results of the last variable in your formula, comparing the
last level of this variable with your base-level. In our case this is
the conditions MOV10_OE vs Control.
results(dds)## log2 fold change (MLE): condition MOV10 OE vs Control
## Wald test p-value: condition MOV10 OE vs Control
## DataFrame with 63345 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue
## <numeric> <numeric> <numeric> <numeric> <numeric>
## ENSG00000000003 3493.2491 -0.4414394 0.0850167 -5.192385 2.07617e-07
## ENSG00000000005 26.2034 -0.0149913 0.4513135 -0.033217 9.73502e-01
## ENSG00000000419 1600.5781 0.3766253 0.1006497 3.741941 1.82605e-04
## ENSG00000000457 504.6637 0.2543571 0.1048510 2.425891 1.52709e-02
## ENSG00000000460 1112.6634 -0.2616540 0.0844934 -3.096739 1.95662e-03
## ... ... ... ... ... ...
## ENSG00000289714 0.000000 NA NA NA NA
## ENSG00000289715 0.000000 NA NA NA NA
## ENSG00000289716 28.595397 0.111404 0.568061 0.196113 0.844522
## ENSG00000289718 0.167597 -1.025123 4.080456 -0.251228 0.801638
## ENSG00000289719 14.922971 0.615131 0.552306 1.113750 0.265387
## padj
## <numeric>
## ENSG00000000003 4.12712e-06
## ENSG00000000005 9.85656e-01
## ENSG00000000419 1.45804e-03
## ENSG00000000457 5.49223e-02
## ENSG00000000460 1.06223e-02
## ... ...
## ENSG00000289714 NA
## ENSG00000289715 NA
## ENSG00000289716 0.915970
## ENSG00000289718 NA
## ENSG00000289719 0.447321However, the dds object stores several results. You can
see these with the function resultsNames().
resultsNames(dds)## [1] "Intercept" "condition_MOV10_KD_vs_Control"
## [3] "condition_MOV10_OE_vs_Control"The Intercept result is a statistical model that compares gene expression to 0 so is not relevant here. We can see that we have results for both of our MOV10 perturbation experiments vs the control.
We can extract a specific result by providing arguments to the
results() function. Let’s look at the first comparison in
our list, MOV10_OE vs Control.
res<-results(dds,contrast = contrasts[[1]])
res## log2 fold change (MLE): condition MOV10_OE vs Control
## Wald test p-value: condition MOV10 OE vs Control
## DataFrame with 63345 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue
## <numeric> <numeric> <numeric> <numeric> <numeric>
## ENSG00000000003 3493.2491 -0.4414394 0.0850167 -5.192385 2.07617e-07
## ENSG00000000005 26.2034 -0.0149913 0.4513135 -0.033217 9.73502e-01
## ENSG00000000419 1600.5781 0.3766253 0.1006497 3.741941 1.82605e-04
## ENSG00000000457 504.6637 0.2543571 0.1048510 2.425891 1.52709e-02
## ENSG00000000460 1112.6634 -0.2616540 0.0844934 -3.096739 1.95662e-03
## ... ... ... ... ... ...
## ENSG00000289714 0.000000 NA NA NA NA
## ENSG00000289715 0.000000 NA NA NA NA
## ENSG00000289716 28.595397 0.111404 0.568061 0.196113 0.844522
## ENSG00000289718 0.167597 -1.025123 4.080456 -0.251228 0.801638
## ENSG00000289719 14.922971 0.615131 0.552306 1.113750 0.265387
## padj
## <numeric>
## ENSG00000000003 4.12712e-06
## ENSG00000000005 9.85764e-01
## ENSG00000000419 1.45804e-03
## ENSG00000000457 5.49223e-02
## ENSG00000000460 1.06223e-02
## ... ...
## ENSG00000289714 NA
## ENSG00000289715 NA
## ENSG00000289716 0.916078
## ENSG00000289718 NA
## ENSG00000289719 0.447321The results table includes several columns:
- baseMean = Mean number of counts from all samples
- log2FoldChange = Log2 of the fold change in normalised counts between sample groups in the contrast
- lfcSE = Standard error of the log2 fold change
- stat = The test statistic (Wald test in this case)
- pvalue = The pvalue / significance level
- padj = The pvalue adjusted for multiple testing
Multiple testing correction and independent filtering
The two most important columns in our results table are log2FoldChange, which is the effect size and tells us how much a gene’s expression has changed, and padj which gives us the level of statistical significance. DESeq2 reports adjusted p-values (padj) which are corrected for multiple testing. We should use these values, not the pvalue column, to filter or call significant genes.
DESeq2 uses the Benjamini-Hochberg method to adjust p-values and control the false discovery rate. So, if you were to filter for genes with a padj<=0.05 you would expect 5% of these to be false positives.
If you inspect the result table you may notice that some genes have
padj and/or pvalue set to NA. This is because the results()
function performs filtering of genes to reduce the total number of genes
tested and increase the likelihood of finding significant genes after
the multiple testing correction. The more genes we test, the larger the
multiple testing correction, so it makes sense to remove genes where we
are unlikely to see a statistical effect:
- Genes with zero counts in all samples
- Genes with extreme outliers
- Genes with extremely low normalised counts
Let’s look at a summary of our results. We will also set an alpha to tell DESeq which significance threshold to use when summarising results:
summary(res,alpha=padj_thresh)##
## out of 36990 with nonzero total read count
## adjusted p-value < 0.05
## LFC > 0 (up) : 2149, 5.8%
## LFC < 0 (down) : 2834, 7.7%
## outliers [1] : 28, 0.076%
## low counts [2] : 18634, 50%
## (mean count < 11)
## [1] see 'cooksCutoff' argument of ?results
## [2] see 'independentFiltering' argument of ?resultsThis is great, we definitely have significant differentially expressed genes!
Log Fold Change Shrinkage
Let’s take a look at these results visually. DESeq2 provides a
plotMA() function to create MA plots(log2FoldChange vs the
mean of normalised counts), a common way to visualise DE genes.
plotMA(res)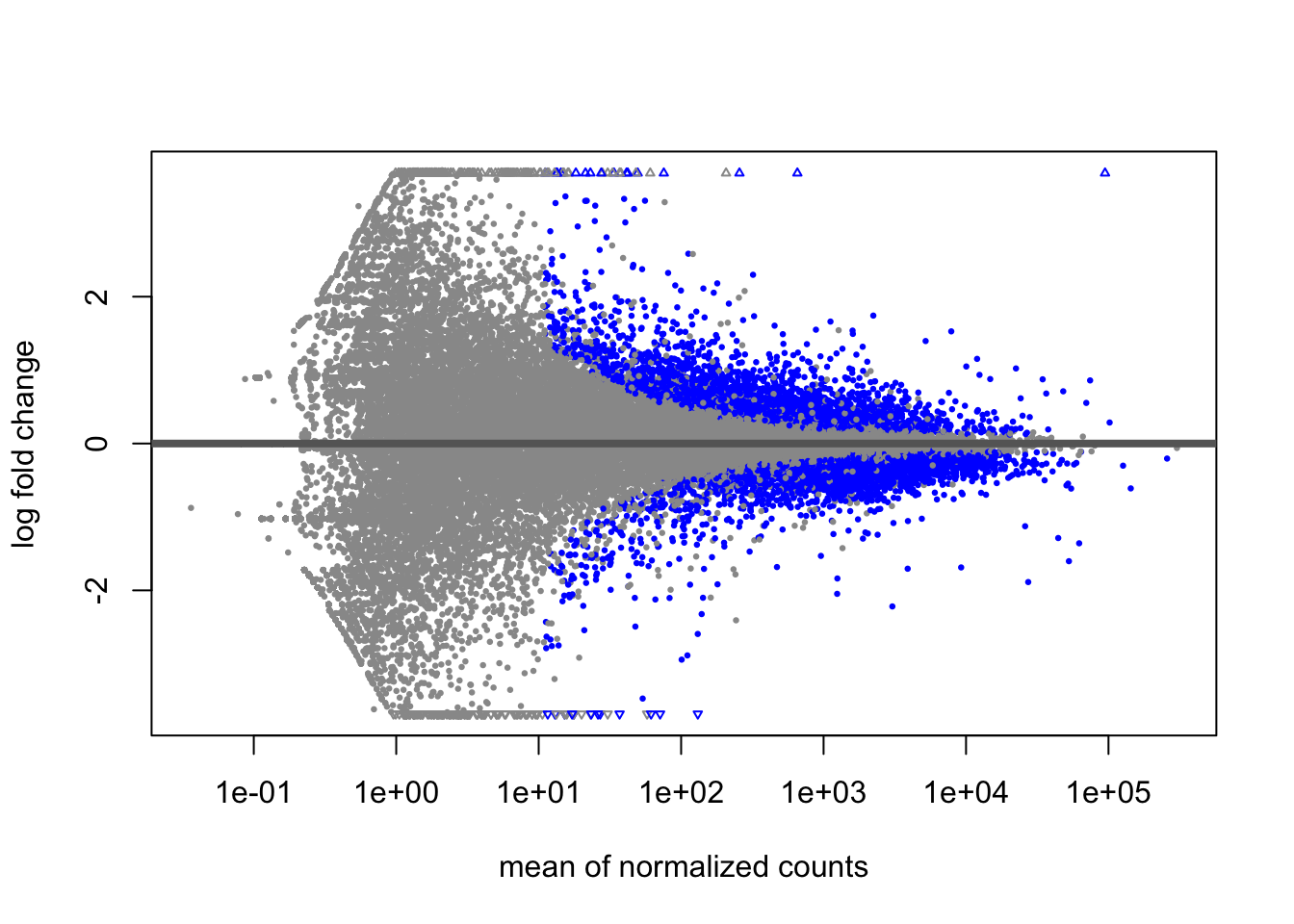
Significant genes (<=0.05 padj) appear in blue, while
non-significant genes are grey. We can immediately see that genes with
low counts have much larger variation in log-fold changes. DESeq2
provides the LFCshrink() to shrink the fold change
estimates and reduce the “noise” from these genes. We can use it instead
of the results() function and these shrunken fold changes
are much better for visualising and ranking our data.
library(apeglm)
resLFC<-lfcShrink(dds,coef = coefficients[1],type="apeglm")## using 'apeglm' for LFC shrinkage. If used in published research, please cite:
## Zhu, A., Ibrahim, J.G., Love, M.I. (2018) Heavy-tailed prior distributions for
## sequence count data: removing the noise and preserving large differences.
## Bioinformatics. https://doi.org/10.1093/bioinformatics/bty895summary(resLFC,alpha=padj_thresh)##
## out of 36990 with nonzero total read count
## adjusted p-value < 0.05
## LFC > 0 (up) : 2149, 5.8%
## LFC < 0 (down) : 2834, 7.7%
## outliers [1] : 28, 0.076%
## low counts [2] : 18634, 50%
## (mean count < 11)
## [1] see 'cooksCutoff' argument of ?results
## [2] see 'independentFiltering' argument of ?resultsWe can see that the number of significant genes is unaffected. Let’s create a new MA plot.
plotMA(resLFC)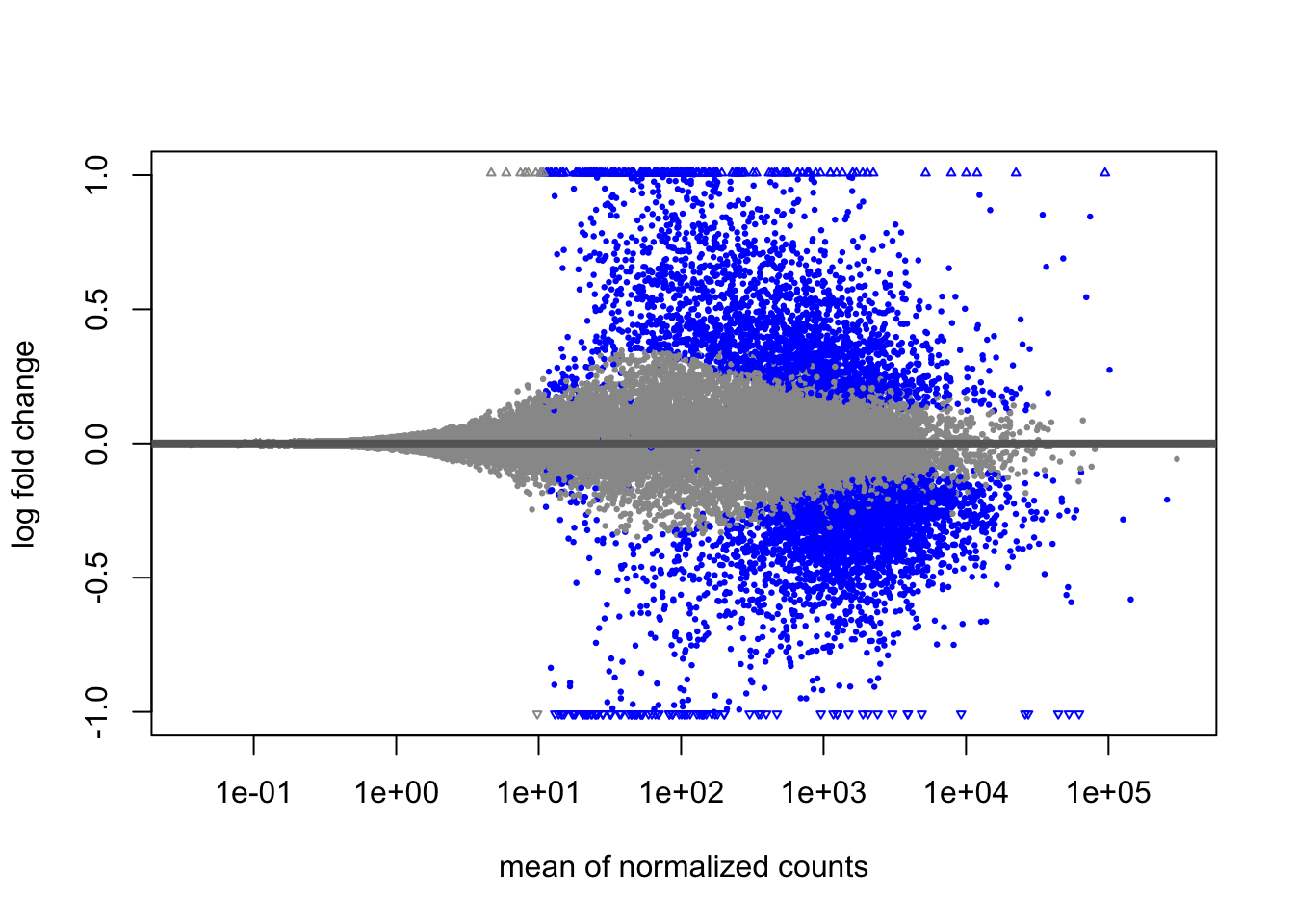
You should see how the shrunken fold changes will be more useful for downstream analysis of the data.
Create result tables for each of our comparisons
Before moving on, we are going to create result tables for each of the comparisons we are interested in. We will also order these by padj so the most significant genes are on top.
## Map each of our coefficients to the lfcShrink function
result_list<-coefficients %>% map(~lfcShrink(dds,coef = .x,type = "apeglm"))## using 'apeglm' for LFC shrinkage. If used in published research, please cite:
## Zhu, A., Ibrahim, J.G., Love, M.I. (2018) Heavy-tailed prior distributions for
## sequence count data: removing the noise and preserving large differences.
## Bioinformatics. https://doi.org/10.1093/bioinformatics/bty895
## using 'apeglm' for LFC shrinkage. If used in published research, please cite:
## Zhu, A., Ibrahim, J.G., Love, M.I. (2018) Heavy-tailed prior distributions for
## sequence count data: removing the noise and preserving large differences.
## Bioinformatics. https://doi.org/10.1093/bioinformatics/bty895names(result_list) = coefficients
result_list## $condition_MOV10_OE_vs_Control
## log2 fold change (MAP): condition MOV10 OE vs Control
## Wald test p-value: condition MOV10 OE vs Control
## DataFrame with 63345 rows and 5 columns
## baseMean log2FoldChange lfcSE pvalue padj
## <numeric> <numeric> <numeric> <numeric> <numeric>
## ENSG00000000003 3493.2491 -0.41896991 0.0857613 2.07617e-07 4.12712e-06
## ENSG00000000005 26.2034 -0.00258219 0.1956445 9.73502e-01 9.85656e-01
## ENSG00000000419 1600.5781 0.34395274 0.1012322 1.82605e-04 1.45804e-03
## ENSG00000000457 504.6637 0.22037898 0.1023773 1.52709e-02 5.49223e-02
## ENSG00000000460 1112.6634 -0.23896825 0.0835393 1.95662e-03 1.06223e-02
## ... ... ... ... ... ...
## ENSG00000289714 0.000000 NA NA NA NA
## ENSG00000289715 0.000000 NA NA NA NA
## ENSG00000289716 28.595397 0.01426524 0.203320 0.844522 0.915970
## ENSG00000289718 0.167597 -0.00642477 0.216954 0.801638 NA
## ENSG00000289719 14.922971 0.08797391 0.224130 0.265387 0.447321
##
## $condition_MOV10_KD_vs_Control
## log2 fold change (MAP): condition MOV10 KD vs Control
## Wald test p-value: condition MOV10 KD vs Control
## DataFrame with 63345 rows and 5 columns
## baseMean log2FoldChange lfcSE pvalue padj
## <numeric> <numeric> <numeric> <numeric> <numeric>
## ENSG00000000003 3493.2491 -0.00474198 0.0832754 0.947750285 0.98066014
## ENSG00000000005 26.2034 -0.07358050 0.1883664 0.204829684 0.43892075
## ENSG00000000419 1600.5781 0.14255488 0.1041493 0.097705231 0.27589567
## ENSG00000000457 504.6637 0.37678932 0.1167932 0.000199266 0.00271642
## ENSG00000000460 1112.6634 0.16833368 0.0886394 0.030134041 0.12505614
## ... ... ... ... ... ...
## ENSG00000289714 0.000000 NA NA NA NA
## ENSG00000289715 0.000000 NA NA NA NA
## ENSG00000289716 28.595397 0.05061218 0.180672 0.298535 0.547702
## ENSG00000289718 0.167597 -0.00326602 0.177681 0.769639 NA
## ENSG00000289719 14.922971 -0.01060597 0.171483 0.817647 0.920636Formatting and annotating results
We will apply a bit of formatting to our results table and also add some annotations.
- Convert to a format where we can use tidyverse verbs
- Move the rownames to a geneID column
- Add a threshold column for genes we wish to label as significant
- Order each table by padj so significant genes are at the top
result_list2<-result_list %>%
map(~as.data.frame(.x) %>%
rownames_to_column("geneID") %>%
mutate(threshold=case_when(padj<=padj_thresh & abs(log2FoldChange)>=l2fc_thresh~"Significant",T~"Not Significant")) %>%
arrange(padj) %>%
as_tibble()
)
result_list2## $condition_MOV10_OE_vs_Control
## # A tibble: 63,345 × 7
## geneID baseMean log2FoldChange lfcSE pvalue padj threshold
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <chr>
## 1 ENSG00000155363 94597. 7.47 0.134 0 0 Significa…
## 2 ENSG00000189060 7875. 1.52 0.0688 1.09e-109 9.95e-106 Significa…
## 3 ENSG00000173110 257. 6.52 0.359 6.20e- 74 3.79e- 70 Significa…
## 4 ENSG00000270882 2234. 1.73 0.101 7.71e- 67 3.53e- 63 Significa…
## 5 ENSG00000187837 1605. 1.48 0.0933 4.53e- 58 1.47e- 54 Significa…
## 6 ENSG00000265972 5204. 1.39 0.0872 4.82e- 58 1.47e- 54 Significa…
## 7 ENSG00000155090 1699. 1.20 0.0805 2.01e- 51 5.26e- 48 Significa…
## 8 ENSG00000102317 8191. -0.750 0.0523 9.57e- 48 2.19e- 44 Significa…
## 9 ENSG00000230678 655. 7.57 0.534 3.50e- 47 7.13e- 44 Significa…
## 10 ENSG00000112972 12430. 0.926 0.0659 4.48e- 46 8.21e- 43 Significa…
## # … with 63,335 more rows
##
## $condition_MOV10_KD_vs_Control
## # A tibble: 63,345 × 7
## geneID baseMean log2FoldChange lfcSE pvalue padj threshold
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <chr>
## 1 ENSG00000116962 6310. 1.10 0.0676 5.57e-61 9.82e-57 Significant
## 2 ENSG00000143183 1784. 1.19 0.0776 2.69e-54 2.37e-50 Significant
## 3 ENSG00000270882 2234. 1.67 0.112 1.82e-51 1.07e-47 Significant
## 4 ENSG00000168036 11650. 0.817 0.0559 7.60e-49 3.35e-45 Significant
## 5 ENSG00000038274 2424. -1.07 0.0748 1.50e-47 5.29e-44 Significant
## 6 ENSG00000116473 2152. -1.02 0.0718 8.37e-47 2.46e-43 Significant
## 7 ENSG00000124762 2759. 1.16 0.0819 1.69e-46 4.25e-43 Significant
## 8 ENSG00000082458 4642. -0.733 0.0535 6.25e-44 1.38e-40 Significant
## 9 ENSG00000109971 22607. 0.739 0.0568 8.34e-40 1.63e-36 Significant
## 10 ENSG00000139219 3768. 0.861 0.0670 7.20e-39 1.27e-35 Significant
## # … with 63,335 more rowsOur result table contains Ensembl gene identifiers but we may want to add more annotations like the gene name and biotype. We can fetch Ensembl annotations from the R package AnnotationHub. Let’s search for the right database first.
library(AnnotationHub)
ah<-AnnotationHub() ## create a link to the annotation hub
query(ah,c("EnsDb","106","Homo")) ##Search annotationHub for Ensembl 106 annotation database## AnnotationHub with 1 record
## # snapshotDate(): 2022-04-21
## # names(): AH100643
## # $dataprovider: Ensembl
## # $species: Homo sapiens
## # $rdataclass: EnsDb
## # $rdatadateadded: 2022-04-21
## # $title: Ensembl 106 EnsDb for Homo sapiens
## # $description: Gene and protein annotations for Homo sapiens based on Ensem...
## # $taxonomyid: 9606
## # $genome: GRCh38
## # $sourcetype: ensembl
## # $sourceurl: http://www.ensembl.org
## # $sourcesize: NA
## # $tags: c("106", "Annotation", "AnnotationHubSoftware", "Coverage",
## # "DataImport", "EnsDb", "Ensembl", "Gene", "Protein", "Sequencing",
## # "Transcript")
## # retrieve record with 'object[["AH100643"]]'You should find the annotation hub ID for the Ensembl v106 humand ensdb object (AH100643). We actually downloaded this earlier with tximeta.
We can then extract gene-level annotations:
edb<-ah[["AH100643"]] ##Load the the ensembldb object## loading from cache## require("ensembldb")## Extract a table of gene annotations
genes<-genes(edb,return.type = "data.frame") %>%
dplyr::select(gene_id,gene_name,gene_biotype,entrezid) %>%
as_tibble()
genesThis looks good but there is something weird about the entrezid column. It actually contains lists of multiple IDs for some Ensembl identifiers. The entrez IDs and Ensembl IDs do not always have a 1-to-1 relationship. To resolve this we will just select the first in each list.
genes$entrezid <- map(genes$entrezid,1) %>% unlist()
genesNow we can merge our result tables with the annotations to add extra columns:
## Map all result lists to a function that joins with the anno data
result_list_anno<-result_list2 %>%
map(~left_join(x=.x,y=genes,by=c("geneID"="gene_id")) %>%
mutate(gene_biotype=as.factor(gene_biotype))) ## biotype as a factorWe will now save these results tables to text files so we can use them outside of R if required. We can create a folder for each of our results.
## Map each of our result names to a function that saves each table in our results_list
names(result_list_anno) %>% map(function(x){
dir.create(x)
write_tsv(result_list_anno[[x]],paste0(x,"/DEseq_result.tsv"),col_names = T,)
})Key points:
- Extract results for individual contrasts
- Create MA plots
- Shrink log2 fold change estimates
- Add annotations to result tables
7. Visualise DESeq2 results
We have already seen the MA plot but there are many other methods for plotting DESeq2 results. We cover some popular visualisations below.
Volcano plots
Let’s try a volcano plot, another popular visualisation for DE analysis. Here we are plotting the log fold change on the x-axis against the negative log of our p-values, so that significant genes appear at the top of the plot. We can then see the spread of fold changes in each direction in our set of significant genes.
ggplot(result_list_anno[[1]],aes(log2FoldChange,-log10(padj),colour=threshold)) +
geom_point() +
theme_bw() +
ggtitle(coefficients[1]) +
theme(legend.key = element_blank())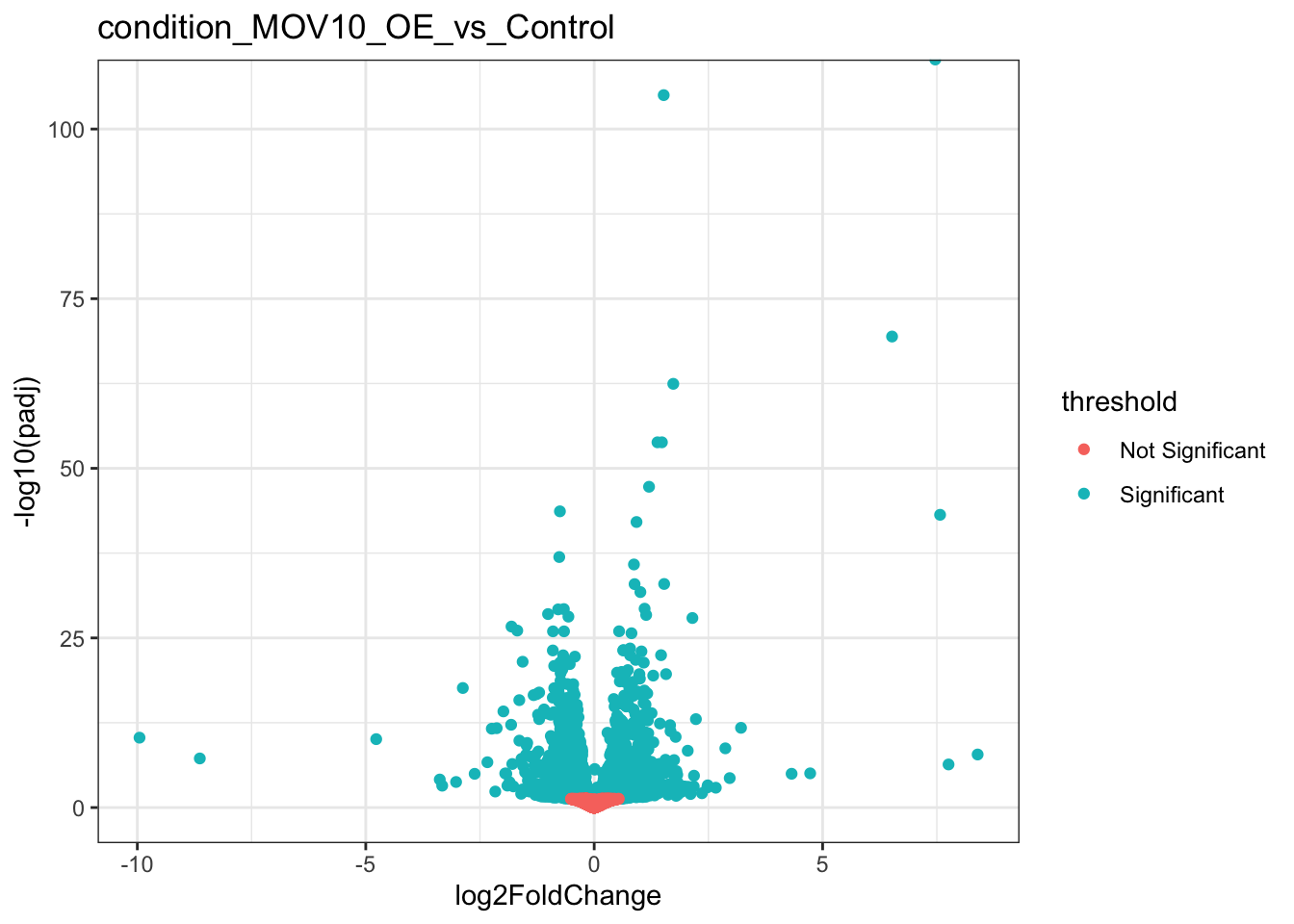
We can even use ggplot to label some of our genes. Let’s select five genes with the lowest adjusted pvalues.
library(ggrepel)
topFive<-result_list_anno[[1]] %>% head(n=5)
ggplot(result_list_anno[[1]],aes(log2FoldChange,-log10(padj),colour=threshold)) +
geom_point() +
geom_text_repel(data=topFive,aes(label = gene_name))+
theme_bw() +
ggtitle(coefficients[1]) +
theme(legend.key = element_blank())## Warning: Removed 45017 rows containing missing values (geom_point).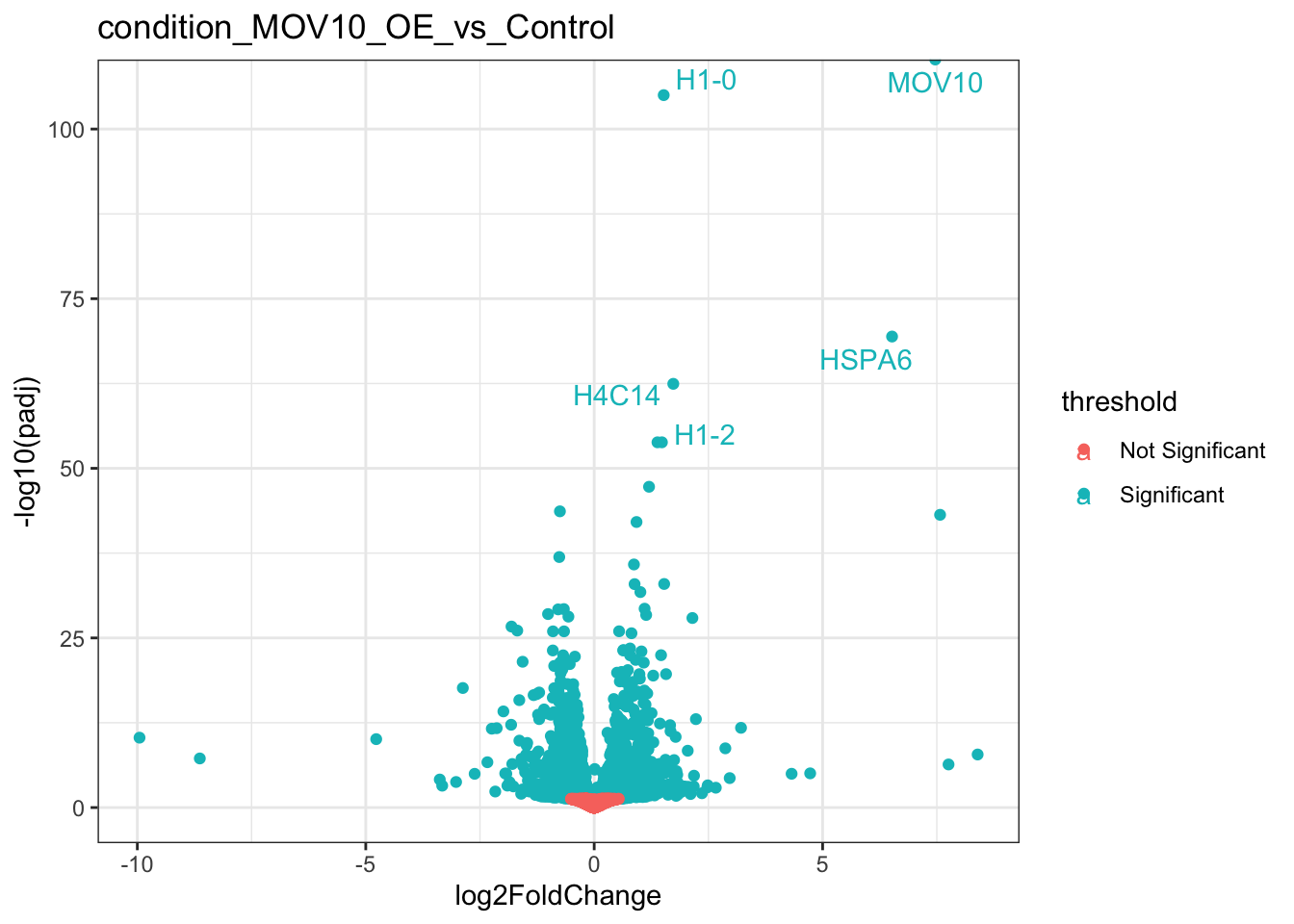
Plots of DE genes by biotype
If we are interested in more than just protein coding genes, we could take a look at the types of RNAs which are represented in our list of DE genes.
First, let’s filter our results for genes which pass our threshold for differential expression.
## Get a list of significant DEGs
sig_genes<-result_list_anno[[1]] %>% dplyr::filter(threshold=="Significant")
## Plot barplot of gene_biotype
ggplot(sig_genes,aes("Significant genes",fill=gene_biotype)) +
geom_bar() +
theme_bw() +
xlab("") +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1))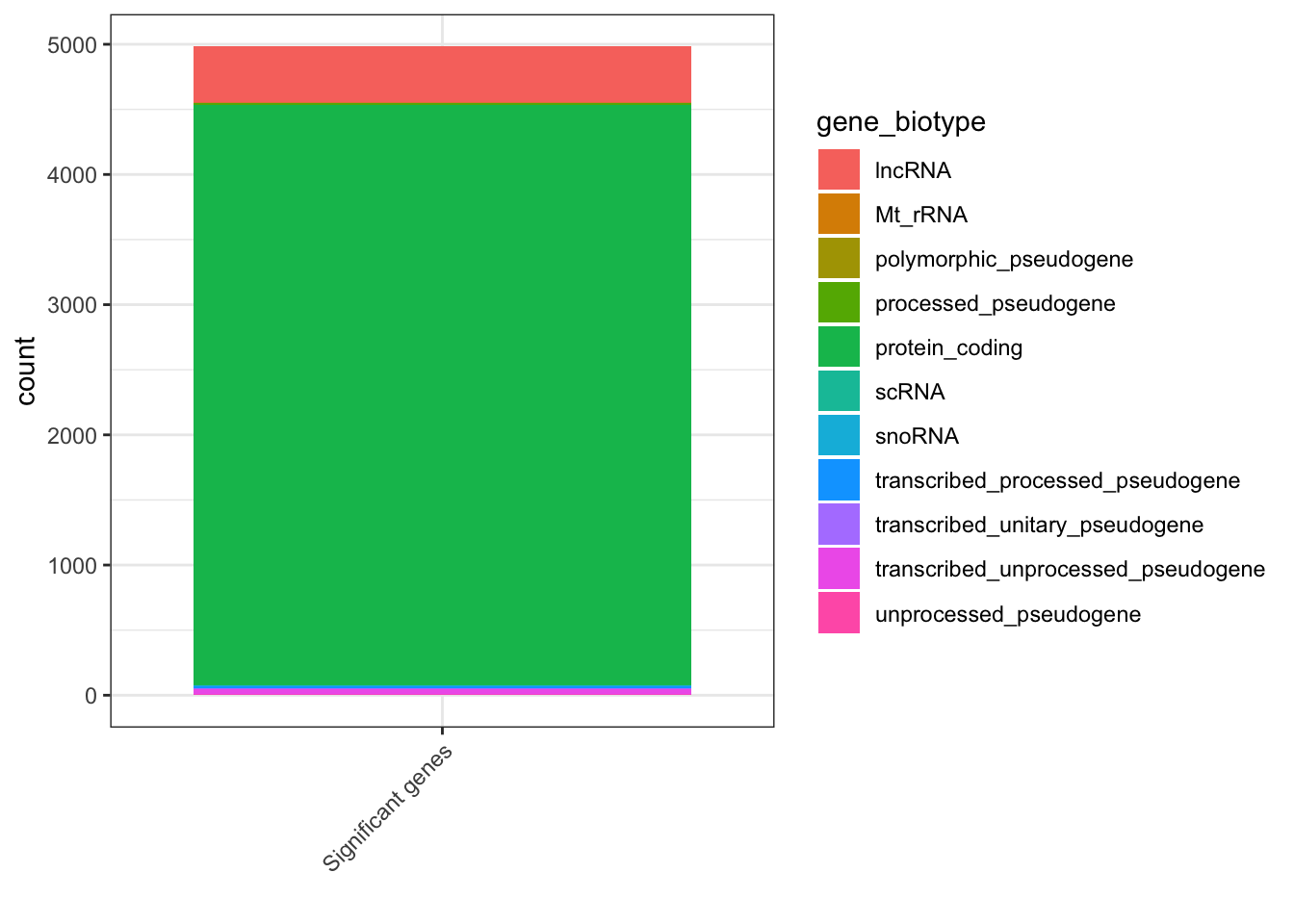
Heatmaps of genes with largest changes
Now that we have our results, we can use the log normalised counts we created earlier to plot heatmaps of genes with the largest changes.
## Out of all significant genes, get the 50 with the largest fold change in either direction in MOV10_OE relative to the control
top50<-result_list_anno[[1]] %>%
dplyr::filter(threshold=="Significant") %>%
arrange(abs(log2FoldChange)) %>%
head(n=50) %>%
pull(geneID)
## Create a matrix of rld counts and plot the heatmap
mat<-assay(rld)[top50,]
colors <- colorRampPalette( rev(brewer.pal(9, "RdBu")) )(255)
pheatmap(mat,color=colors,scale = "row",cluster_rows = T,cluster_cols = T,annotation_col = anno)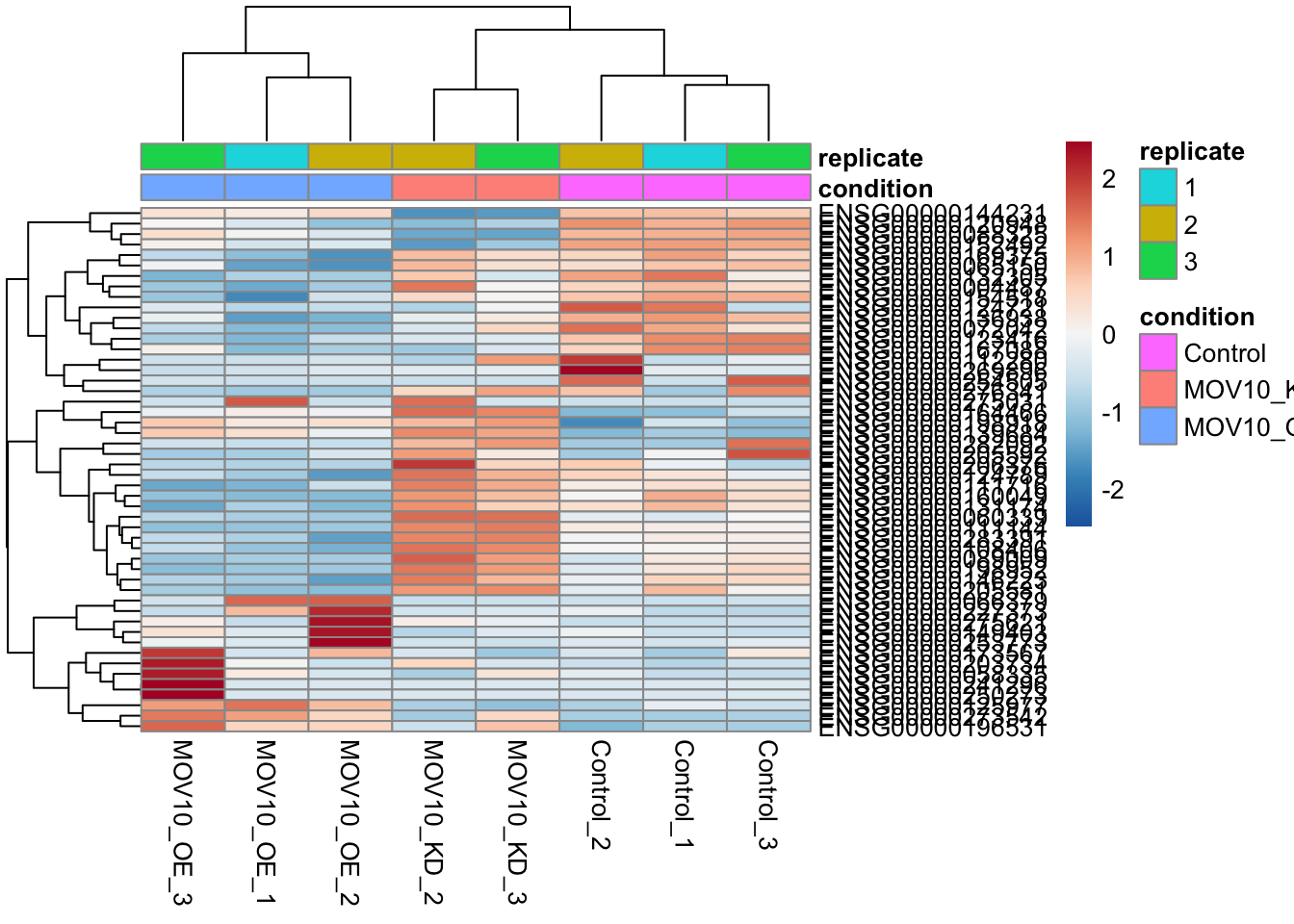
Key points:
- There are many ways to summarise and visualise DE results
- Get creative!
8. Functional enrichment of genes
We can perform enrichment analyses to see if there are any functional annotations common to our DE genes that may imply something about the function of MOV10.
In these examples we will use all of our significant genes. However, in some cases it may be more appropriate to look at genes that are differentially expressed in one direction.
The clusterProfiler package has functions for enrichment analysis and plotting gene annotations. To perform the enrichment analysis, we need a list of background genes and a list of significant genes. For our background dataset we will use all genes tested for differential expression (all genes in our results table).
The enrichGO function looks for gene ontology terms that are over-represented in the list of significant genes. We will look at the Biological Process (BP) terms only.
library(clusterProfiler)
library(org.Hs.eg.db)
## Run the GO enrichment
ego <- enrichGO(gene = sig_genes$geneID,
universe = result_list_anno[[1]]$geneID,
OrgDb = org.Hs.eg.db,
keyType = "ENSEMBL",
ont = "BP",
pAdjustMethod = "BH",
qvalueCutoff = 0.05,
readable = TRUE)
head(ego)## Plot the top enriched terms
dotplot(ego, showCategory=20)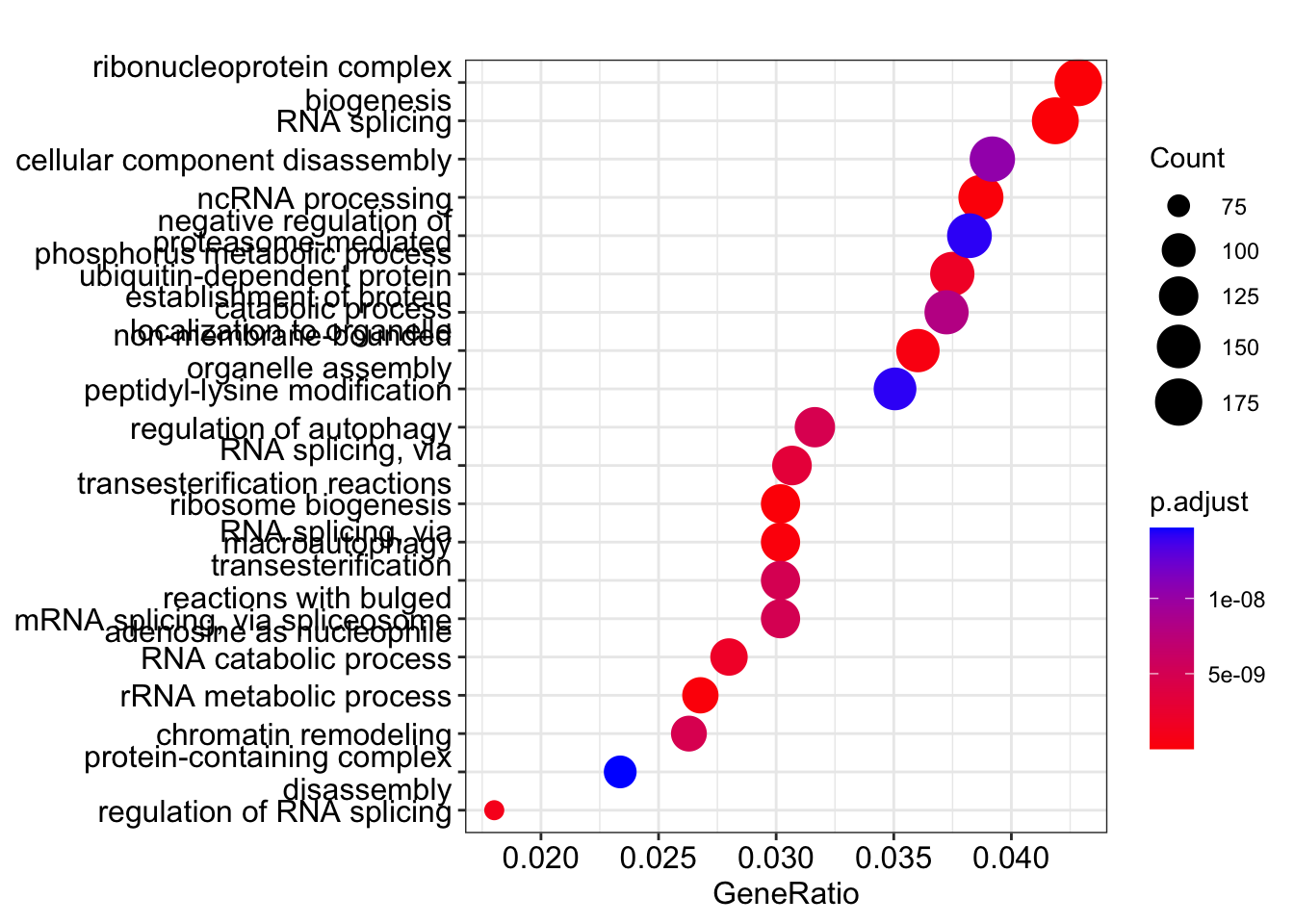
Further Learning
clusterProfiler also provides methods to query pathway databases such as KEGG. You can find out more in the documentation.
g:Profiler
The package gprofiler2 is a wrapper for the online tool g:Profiler and looks at many different databases at once including GO, KEGG and transcription factor motifs. g:Profiler performs functional enrichment analysis of gene lists using Ensembl identifiers.
library(gprofiler2)
## Get a table of enriched terms with the gost function - this accepts Ensembl gene IDs
gp<-gost(query = sig_genes$geneID,organism = "hsapiens", ordered_query = FALSE,multi_query = FALSE, significant = TRUE, exclude_iea = TRUE, user_threshold = 0.05, correction_method = "g_SCS", domain_scope = "annotated")
## Inspect the output, the "term_name" column is the enriched term and the "source" tells us which database it belongs to.
gp$resultKey points:
- Bioconductor has many packages for functional enrichment analysis
- Find annotation terms that are statistically over-represented in lists of significant DEGs
- Test for terms in genes going up, down and in either direction relative to the reference sample
Challenge:
See if you can produce similar visual outputs and summaries of results for the MOV10_KD_vs_Control comparison.